Dr. Christiane Ring, Dr. Bastian Thaa, Dr. Marc Esser, Dr. Carsten Schwenke (veröffentlicht in Market Access & Health Policy 2019; 9(6):26–28)
Surrogate in der frühen Nutzenbewertung
Die Validierung bleibt eine Herausforderung
Die Anforderungen der Zulassungsbehörden unterscheiden sich von denen der frühen Nutzenbewertung in Deutschland. Die Zulassung von Arzneimitteln kann auch auf klinischen Studien basieren, die im Rahmen des Wirksamkeitsnachweises hauptsächlich Surrogate anstelle von patientenrelevanten Endpunkten untersucht haben. Hingegen zieht der G-BA nur patientenrelevante Endpunkte heran, die vom Patienten spürbar sind, oder jene Surrogate, für die durch Validierung belegt wurde, dass damit Rückschlüsse auf patientenrelevante Endpunkte möglich sind.
Surrogate ersetzen patientenrelevante Endpunkte
Surrogate sind physiologische oder biochemische Marker, die für den Patienten in der Regel nicht direkt spürbar sind, sondern stellvertretend für wichtige klinische Endpunkte bestimmt werden. Sie müssen im pathophysiologischen Zusammenhang mit dem eigentlichen patientenrelevanten Endpunkt stehen und eine Prognose desselben ermöglichen. Surrogate werden in klinischen Studien häufig eingesetzt, weil damit Veränderungen im Krankheitsverlauf früher erkannt und besser quantifiziert werden können. So wird zum Beispiel die Konzentration der alkalischen Phosphatase zusammen mit dem Gesamtbilirubinwert als Surrogat für die Leberfunktion verwendet. Während für die Zulassung eines Arzneimittels solche Endpunkte ausreichen können, sind die Anforderungen für die Nutzenbewertung höher: Damit Surrogate in der frühen Nutzenbewertung von G-BA und IQWiG berücksichtigt werden können, müssen sie validiert sein, das bedeutet, die Korrelation zwischen den Effekten der Intervention auf das Surrogat und den patientenrelevanten Endpunkt, den das Surrogat ersetzen soll, muss nachgewiesen sein.
Eine Handvoll anerkannter Surrogate
Etwa jedes vierte Nutzendossier setzt Surrogate ein, aber nur in den wenigsten Dossiers wird tatsächlich ein Validierungsversuch vorgenommen.1 Meistens werden die eigentlichen Surrogate stattdessen als patientenrelevante Endpunkte im Dossier dargestellt oder – wenn sie als Surrogate genannt werden – lediglich mit Argumenten gestützt. Entsprechend wird die überwiegende Mehrheit der Surrogate in der Nutzenbewertung des IQWiG nicht berücksichtigt und im Beschluss des G-BA gar nicht oder lediglich ergänzend dargestellt. Nur in wenigen Einzelfällen wurden Surrogate bisher als valide und patientenrelevant anerkannt (Tab. 1).
Zu den bisher vom G-BA akzeptierten Surrogaten gehören das virologische Ansprechen – auch als Viruslast bezeichnet – und die CD4-Zellzahl als Surrogate für das Auftreten von AIDS-definierenden Erkrankungen bzw. Tod bei Infektion mit HIV. Bereits 2012 stufte das IQWiG in der Nutzenbewertung von Rilpivirin die Viruslast basierend auf Hughes 20052 als ausreichend valides Surrogat ein, jedoch mit erhöhter Unsicherheit hinsichtlich des Zusatznutzens, da für die deutliche Korrelation der Effekte keine statistische Signifikanz gegeben war. Auch die Validität der CD4-Zellzahl begründete das IQWiG 2013 anhand dieser Publikation in der Nutzenbewertung der Elvitegravir-Fixkombination. Seither wird in Nutzendossiers in dieser Indikation in der Regel darauf verwiesen, dass die genannten Surrogate bereits anerkannt worden sind.
Die Reduktion oraler Glukokortikoide (OCS) wurde vom G-BA als Surrogat für die Vermeidung Glukokortikoid-induzierter Nebenwirkungen in den beiden Indikationen Asthma und systemischer Lupus erythematodes (SLE) eingestuft und mit der Begründung akzeptiert, dass die OCS-Therapie direkt mit Nebenwirkungen assoziiert ist und die Vermeidung einer solchen Therapie daher ein wichtiges Therapieziel darstellt. In den entsprechenden Nutzendossiers war keine Validierung vorgelegt, sondern die OCS-Reduktion als patientenrelevanter Endpunkt dargestellt worden. Das IQWiG kommentierte die OCS-Reduktion jeweils nicht, weil insgesamt für die Nutzenbewertung keine relevanten Daten vorlagen.
Der HbA1c-Wert dient als klinischer Langzeitmarker für durchschnittliche Blutzuckerwerte und wird bei Diabetes-Patienten routinemäßig gemessen. Bei Diabetes mellitus Typ 1 wurde im Verfahren zu Insulin degludec 2015 der HbA1c-Wert von G-BA und IQWiG als valides Surrogat für mikrovaskuläre Folgekomplikationen anerkannt, basierend auf zwei Publikationen zur Validität. Der HbA1c-Wert war im Nutzendossier ursprünglich nur unterstützend dargestellt worden. Bei Diabetes Typ 2 hingegen erachteten G-BA und IQWiG den HbA1c-Wert weder als valides Surrogat noch als per se patientenrelevant und stellten entsprechende Daten in der Nutzenbewertung bzw. im Beschluss lediglich ergänzend dar.
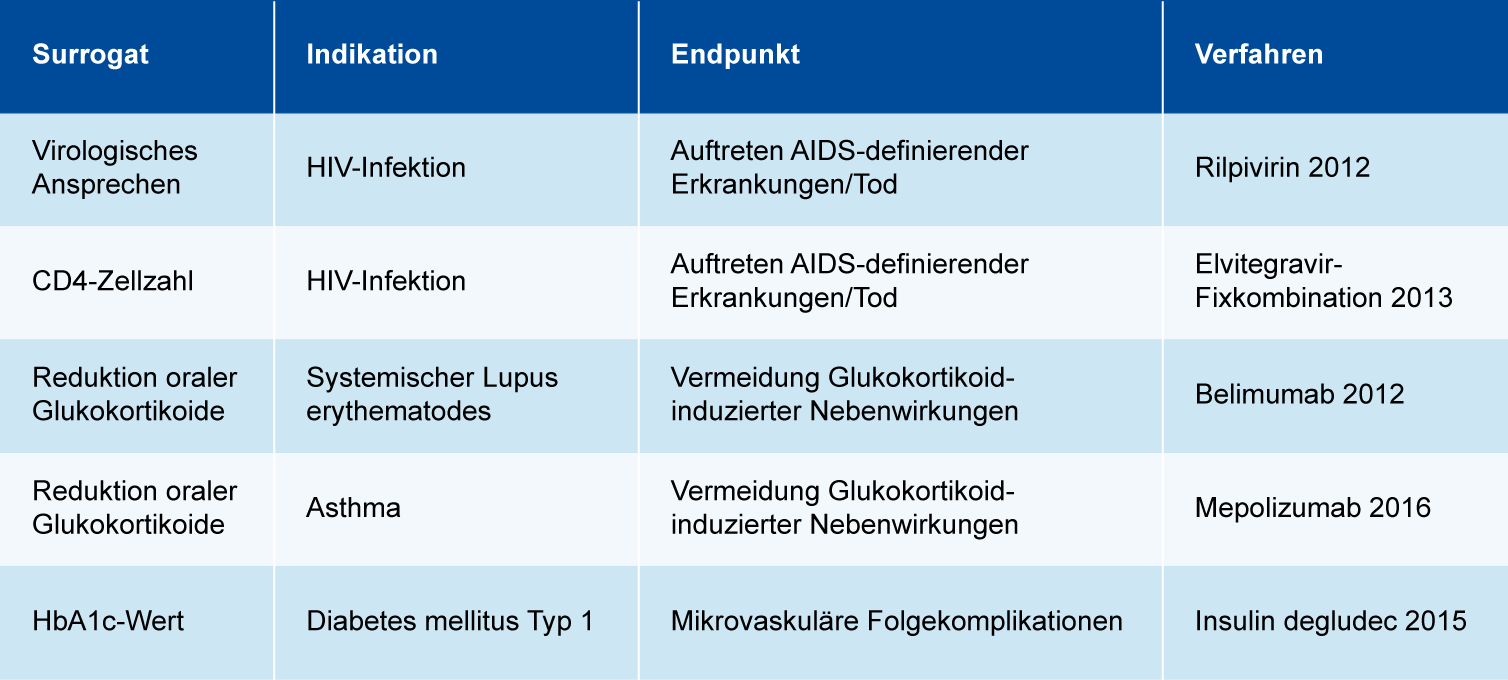
Tab. 1: Übersicht über die Surrogate, die bisher in einem Verfahren der frühen Nutzenbewertung vom G-BA akzeptiert wurden, mit Angabe der jeweilige Indikation, dem entsprechende patientenrelevanten Endpunkt und des Verfahren, in dem das Surrogat erstmals akzeptiert wurde.
Was eine erfolgreiche Validierung ausmacht
Damit ein Surrogat anstelle eines patientenrelevanten Endpunktes betrachtet werden kann, müssen drei Aspekte erfüllt sein (Abb. 1). Zum einen muss das Surrogat biologisch plausibel sein. Es muss also einen kausalen pathophysiologischen Zusammenhang zwischen dem Surrogat und dem eigentlichen patientenrelevanten Endpunkt geben. Die biologische Plausibilität lässt sich mit Erkenntnissen aus der Grundlagenforschung begründen. Des Weiteren muss eine Korrelation zwischen Surrogat und Endpunkt vorliegen. Veränderungen des klinischen Endpunktes muss das Surrogat sowohl qualitativ als auch quantitativ widerspiegeln. Epidemiologische Studien sind geeignet, um solche Zusammenhänge aufzuzeigen. Schließlich muss die Intervention auf das Surrogat den gleichen Effekt wie auf zu ersetzenden Endpunkt haben. Dieses Kriterium kann nur mit Ergebnissen aus randomisierten kontrollierten Studien belegt werden.3

Abb.1: Valide Surrogate müssen drei Anforderungen erfüllen. Abbildung basierend auf 3.
Im IQWiG-Bericht 2011 zu Surrogaten in der Onkologie4 werden korrelationsbasierte Validierungsverfahren als praktikabel vorgeschlagen. Wichtig ist dabei, dass die Validierung krankheits- und indikationsspezifisch erfolgt und die eingeschlossenen Studien vergleichbar sind. Eingeschlossen werden können dabei nur Studien, die Daten zu sowohl dem Surrogat als auch dem eigentlichen Endpunkt gesammelt haben. Bei mindestens mittlerer Korrelation kann mit Hilfe des Surrogate Threshold Effects (STE) statistisch ermittelt werden, ab welchem Schwellenwert vom Behandlungseffekt auf das Surrogat auf einen Effekt im eigentlich interessierenden Endpunkt geschlossen werden kann. Mit der Dokumentation der Validierung im Nutzendossier prüft das IQWiG die Validität des Surrogats hinsichtlich ihrer Aussagesicherheit und passt die Sicherheit seiner Bewertung des patientenrelevanten Endpunktes gegebenenfalls an.
Für das mit diagnostischer Bildgebung ermittelte (d.h. nicht auf Symptomerfassung basierende) PFS als Surrogat für den Endpunkt Gesamtüberleben in der Onkologie wurden in der Vergangenheit bereits mehrfach Validierungsversuche in Nutzendossiers dargestellt. Im Nutzenbewertungsverfahren zu Dabrafenib in der Indikation Melanom von 2014 verwendete der pharmazeutische Unternehmer für die Korrelationsanalyse ein einfaches lineares Modell. Dieser methodische Ansatz wurde vom IQWiG nicht akzeptiert, da er die Variabilität der Effektschätzer nicht berücksichtigt. Zudem legte das IQWiG in seiner Bewertung dar, dass die für die Nutzenbewertung relevante Studie nicht für die Validierung herangezogen werden könne und dass bei heterogenen Interventionen in den eingeschlossenen Studien auch der Einfluss verschiedener Therapiearten untersucht werden müsse. In der Indikation Melanom wurde im Jahr 2016 im Nutzenbewertungsverfahren zu Pembrolizumab erneut ein Validierungsversuch für das PFS vorgelegt. Diesmal wurde der prinzipiell geeignete STE-Ansatz verwendet, allerdings wurden vom IQWiG die Wahl der Population und Intervention sowie das Fehlen einer Registerrecherche kritisiert. Auch in der Indikation Brustkrebs wurde 2018 im Verfahren zu Palbociclib die STE-Analyse zur Validierung des PFS herangezogen. Hier waren ebenfalls die Wahl der eingeschlossenen Studien bezüglich der Intervention und eine fehlende Registerrecherche Kritikpunkte des IQWiG. Hinzu kamen eine aus Sicht des IQWiG fehlerhafte Methodik und der Einschluss der Zeit bis zur Progression, die jedoch nicht als äquivalent zum PFS angesehen werden kann.
Vom IQWiG generell positiv beurteilt wurde der Validierungsversuch des krankheitsfreien Überlebens (DFS) als Surrogat für das Gesamtüberleben, der 2018 im Nutzenbewertungsverfahren zu Pertuzumab vorgelegt wurde. Darin eingeschlossen wurden acht Studien, die jeweils einen Interventionsarm mit einem Arzneimittel aus derselben Wirkstoffklasse wie Pertuzumab und mit einer der Zulassung von Pertuzumab entsprechenden Verabreichung umfassten. Die Zielpopulation von Pertuzumab wurde in einer Subgruppenanalyse erfasst. Das IQWiG erachtete eine Assoziation auf Studienebene als entscheidend, dabei zeigte ein lineares Regressionsmodell unter Gewichtung der Todesfälle jeder Studie eine mittlere Korrelation der Effekte. Die uneingeschränkte Validität des Surrogats hätte nur mit einer hohen Korrelation (definiert als untere Grenze des 95-%-Konfidenzintervalls mindestens 0,85) gezeigt werden können. Daher wurde ein STE berechnet und mit den Effektschätzern der zu bewertenden Pertuzumab-Studie verglichen. Da das Konfidenzintervall des DFS nicht vollständig unterhalb des STE lag, konnte von den Effekten von Pertuzumab auf das DFS nicht auf einen relevanten Effekt auf das Gesamtüberleben geschlossen und entsprechend ein Zusatznutzen nicht gezeigt werden. Nichtdestotrotz wurde das Vorgehen bei der Validierung vom IQWiG als geeignet anerkannt.
Fazit: Strenge Anforderungen an die Validierung – unerfüllbar?
Pharmazeutische Unternehmer stehen bei der Erstellung des Nutzendossiers vor der großen Herausforderung, dass in vielen Fällen Daten zum eigentlich interessierenden patientenrelevanten Endpunkt nicht verfügbar sind und sie auf Surrogate zurückgreifen müssen. Gleichzeitig fehlt es jedoch auch an geeigneten Studien, um das entsprechende Surrogat zu validieren. So gehören unter anderem ungeeignete Patientenpopulationen oder Interventionen zu den Gründen, warum Validierungen scheitern.
Die Akzeptanz von Surrogatvalidierungen wird zukünftig nur dann gesteigert werden können, wenn wie im Nutzendossier zu Pertuzumab die strengen Richtlinien des IQWiG strikt eingehalten werden. Auch wenn alle Formalien erfüllt sind, werden jedoch weiterhin für viele Validierungen die geeigneten Studiendaten fehlen. Die Generierung von geeigneten Studiendaten unter Berücksichtigung der Anforderungen an eine Surrogatvalidierung in Deutschland wäre wünschenswert, ist in internationalen Studien aber kaum umsetzbar.
Referenzen
1 Ring C et al. PNS263: The role of surrogates for patient-relevant endpoints in the early benefit assessment in Germany. 2019. Poster ISPOR Europe, Copenhagen.
2 Hughes MD. In: Burzykowski T, Molenberghs G, Buyse M (Ed). The evaluation of surrogate endpoints. New York: Springer; 2005. S. 295-321.
3 Mangiapane S, Velasco Garrido M. Surrogatendpunkte als Parameter der Nutzenbewertung. Schriftenreihe Health Technology Assessment, Bd. 91. Herausgegeben vom DIMI, Köln 2009. https://portal.dimdi.de/de/hta/hta_berichte/hta250_bericht_de.pdf [Zugriff: 08.10.2019].
4 IQWiG. Rapid Report A 10-05 Aussagekraft von Surrogaten in der Onkologie. 2011. https://www.iqwig.de/download/A10-05_Rapid_Report_Version_1-1_Surrogate_in_der_Onkologie.pdf [Zugriff: 08.10.2019].